La enfermedad de Huntington, también conocida como Corea de Huntington, es un trastorno neurológico degenerativo y hereditario. Esta enfermedad afecta tanto a hombres como a mujeres.
Se trata de una patología causada por el desgaste neuronal en ciertas zonas cerebrales. Lo más habitual es que la enfermedad de Huntington aparezca en edad adulta, pero también hay casos que se detecta en la adolescencia.
Aquellas personas que padezcan corea de Huntington pueden tener un bebé, aunque es importante recibir previamente asesoramiento genético. Una opción sería recurrir a la fecundación in vitro con diagnóstico genético preimplantacional para evitar la transmisión de la enfermedad a la descendencia.
A continuación tienes un índice con los 9 puntos que vamos a tratar en este artículo.
- 1.
- 1.1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 6.1.
- 6.2.
- 6.3.
- 6.4.
- 7.
- 8.
- 9.
¿Qué es la corea de Huntington?
La corea de Huntington, enfermedad de Huntington o corea progresiva crónica es una patología neurodegenerativa y hereditaria que se transmite de padres a hijos. El nombre de esta enfermedad es en honor a un médico estadounidense que la descubrió en 1872, el Dr. George Huntington.

La forma más habitual de esta enfermedad es la que aparece en la edad adulta.
Lo que ocurre es que los pacientes nacen con la alteración genética, pero no es hasta los 30-40 años cuando surgen las manifestaciones clínicas típicas de la enfermedad de Huntington.
La característica más destacada de la enfermedad es la degeneración neuronal en varios puntos del cerebro, por lo que habrá una afectación a nivel de movilidad y coordinación.
¿Qué síntomas provoca?
Al inicio de la enfermedad, los síntomas suelen ser sutiles. Sin embargo, en la edad adulta, entorno a los 30-40 años, las manifestaciones clínicas de la enfermedad de Huntington suelen ser más notorias.
A continuación, se detallan cada uno de los signos propios de la corea de Huntington:
- Corea o movimientos involuntarios
- se trata de movimientos rápidos y bruscos que ocurren sin ninguna causa aparente. Este tipo de movimientos puede surgir en distintas zonas del cuerpo como, por ejemplo, las piernas, los brazos o incluso en la cara.
- Bradicinesia
- a diferencia de la corea, la bradicinesia consiste en movimientos voluntarios que se realizan lentamente.
- Cambios de comportamiento y personalidad
- ese suele ser uno de los primeros síntomas de la corea de Huntington. La apatía, la irritabilidad o ser impulsivos son algunas de las características de esta enfermedad.
- Pérdidas de habilidades cognitivas
- las personas que padecen la enfermedad de Huntington suelen presentar dificultad para concentrarse, falta de memoria, etc.
- Ansiedad y depresión
- los problemas a nivel psiquiátrico también es uno de los síntomas de la enfermedad.

Aparte de todos estos síntomas, también se ha visto dificultad en el habla, problemas en la deglución de alimentos, rigidez muscular, etc. No obstante, estos síntomas asociados a la enfermedad de Huntigton pueden variar de una persona a otra y también en función del grado de severidad, velocidad en la que progresa la enfermedad, etc.
Causas de la enfermedad de Huntington
Se trata de una enfermedad monogénica, es decir, la enfermedad de Huntington está causada por alteraciones de un único gen (el gen HTT). Además, su herencia es autosómica dominante, lo que significa que únicamente es necesario una copia del gen alterado para que el individuo presente corea de Huntington.
Pese a que, la mayoría de veces, la corea de Huntington es heredada, existen un pequeño porcentaje de casos donde la enfermedad aparece de novo.
El gen causante de la enfermedad de Huntington se encuentra en el brazo corto del cromosoma 4 y codifica una proteína conocida como la huntingtina. Cuando se produce una mutación del gen, lo que sucede es que hay más repeticiones del triplete CAG en el gen HTT de las que debería.
Una persona sana presenta 10-27 repeticiones del triplete CAG; mientras que si ha heredado la mutación causante de la enfermedad de Huntington presentará 36-120 repeticiones. En el caso de que únicamente sea portadora de la enfermedad, tendrá entre 27 y 35 repeticiones del triplete.
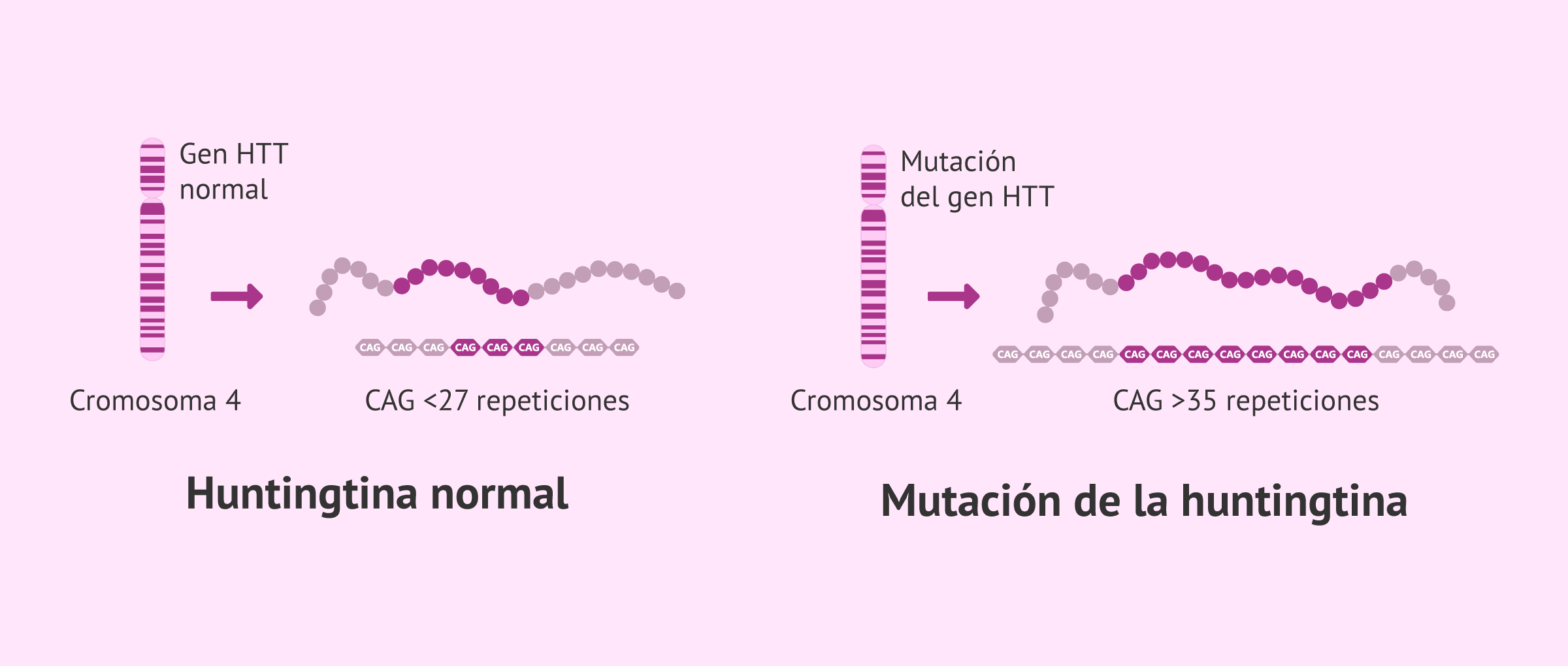
Cabe destacar que, a medida que la mutación del gen se va transmitiendo a la descendencia, el número de repeticiones del triplete CAG aumenta. Por ello, es posible que los síntomas de la enfermedad de Huntington aparezcan cada vez más pronto.
¿Cómo se diagnostica la enfermedad de Huntington?
El diagnóstico de la enfermedad de Huntington comienza con una exploración física donde se intenta detectar síntomas motores, sensoriales y psiquiátricos, así como un análisis neuropsicológico donde se evalúan aspectos relacionados con la memoria, el razonamiento, la agilidad mental, etc. Además, es importante conocer los antecedentes familiares.
Conocer los antecedentes familiares es fundamental para el diagnóstico de un posible caso de Corea de Huntington.
Si hay sospechas de corea de Huntington, el especialista solicitará pruebas complementarias de imagen, tales como una tomografía computarizada o una resonancia magnética. Las imágenes del cerebro obtenidas a través de estas pruebas informará al especialista de posibles cambios detectados y descartará otras patologías que podrían ser causas de los síntomas manifestados.

La confirmación del diagnóstico de la corea de Huntington es a través de una prueba genética. Para ello, únicamente se tomará una muestra de sangre del paciente y se extraerá su ADN para estudiar el número de repeticiones del triplete CAG en el gen que codifica para la Huntingtina.
Tratamiento
La enfermedad de Huntington no tiene cura definitiva, pero sí existen diferentes terapias para controlar los síntomas, aunque no para frenar el deterioro neurológico que ocasiona este trastorno.
A continuación, se detallan algunas de las terapias para aliviar las manifestaciones clínicas de la enfermedad de huntington:
- Uso de medicamentos como benzodiazepinas o tetrabenazina que ayudan a controlar la corea, es decir, los movimientos involuntarios y espasmódicos. Además, también se indican los antidepresivos o estabilizadores de ánimo, entre otros fármacos.
- Psicoterapia para abordar los problemas de comportamiento.
- Fisioterapia enfocada a mejorar la fuerza de los músculos, la coordinación en los movimientos, etc.

Además, se puede realizar terapia del habla con ayuda de un logopeda puesto que la enfermedad de Huntignton puede afectar al habla y/o a la deglución.
Enfermedad de Huntington y embarazo
Gracias a los avances en medicina, las personas portadoras de la enfermedad de Huntington pueden tener descendencia sin transmitir la mutación. Para ello, deberán recurrir a las técnicas de reproducción asistida, en concreto, a la fecundación in vitro con diagnóstico genético preimplantacional (FIV con DGP).
La mayoría de pacientes cometen el mismo error a la hora de elegir clínica de reproducción asistida.
Si accedes a nuestra guía Fertilidad con Cabeza te explicaremos cuál es el error más común para que puedas evitarlo.
En este caso, la fecundación ocurre en el laboratorio y los embriones generados son analizados genéticamente. De este modo, los embriones alterados genéticamente serán descartados, transfiriéndose únicamente los embriones sanos y, por tanto, sin la mutación del gen de la Huntingtina.
Además de la FIV con DGP, también existe la opción de hacer la técnica de amniocentesis una vez logrado el embarazo. Sin embargo, esta prueba prenatal invasiva supone más riesgos para el feto.
Preguntas de los usuarios
¿La enfermedad de Huntington afecta a la fertilidad?
La enfermedad de Huntington es un trastorno neurológico hereditario poco común que afecta el sistema nervioso central. Se hereda a través de una mutación genética específica. Dado que es una enfermedad genética, es comprensible que se generen preocupaciones sobre sus efectos en la salud reproductiva, incluida la fertilidad.
En términos de impacto directo en la fertilidad, la enfermedad de Huntington en sí misma no se asocia con una disminución de la fertilidad. Las personas con esta enfermedad, en general, pueden concebir de manera natural. Los problemas de fertilidad relacionados con esta enfermedad suelen ser secundarios y vinculados a las complicaciones neurológicas y de salud en etapas posteriores de la enfermedad. Por lo tanto, la capacidad reproductiva inicial generalmente no se ve directamente afectada.
Sin embargo, es fundamental considerar varios aspectos cuando se trata de planificar la familia en casos de enfermedad de Huntington. El mayor dilema reside en la transmisión de esta afección a la descendencia. Dado que es un trastorno genético autosómico dominante, existe un 50% de probabilidad de transmitir el gen mutado a los hijos. Esto plantea una preocupación legítima para las parejas en las que uno o ambos padres son portadores del gen de la enfermedad.

En estos casos, muchas parejas recurren al asesoramiento genético y las pruebas genéticas preconcepcionales para determinar el riesgo de transmitir la enfermedad a su descendencia. Dependiendo del resultado, pueden optar por la fertilización in vitro (FIV) con diagnóstico genético preimplantacional (DGP) para seleccionar embriones que no porten la mutación. Esto permite evitar que la enfermedad se transmita a la descendencia.
¿Puedo ser madre si sufro la enfermedad de Huntington?
La enfermedad de Huntington es una patología hereditaria que se caracteriza por debutar a edad adulta. En muchos casos, cuando el enfermo debuta y es diagnosticado con esta patología ya ha tenido decencia de forma natural. Es por ello que la mayoría de casos que se presentan en reproducción asistida es de pacientes de segunda generación con antecedentes de esta enfermedad.
La forma de proceder más común es mediante un tratamiento de Fecundación In Vitro que permita al laboratorio realizar un Diagnóstico Genético. Mediante esta técnica se puede detectar aquellos embriones que presentan la enfermedad y descartaros. Sólo aquellos embriones libres de la enfermedad serán transferidos a la paciente.
Otra de las posibilidades que se presentan es la donación de gametos donados (óvulos o espermatozoides), de esta forma se elimina la posibilidad de heredar la patología.
La enfermedad de Huntintong presenta un reto para aquellos pacientes con antecedentes familiares de la enfermedad que deseen ser padres de un hijo sano pero que no quieran saber si ellos son portadores de la enfermedad.
Para ello se ha desarrollado un sofisticado protocolo de actuación para evitar la transmisión de la enfermedad a los embriones creados por FIV y, a la vez, mantener el derecho del parental con posibilidad de ser portador a no querer ser informado de las posibilidades de tener el gen de la enfermedad de Huntington. De esta manera se analiza los genes del parental con posibilidades de estar afectado por la enfermedad, y si procede, se realiza el Diagnóstico Genético a los embriones. Como resultado, se cercioran que los embriones a transferir son totalmente sano y libre de la enfermedad de Huntington a la vez que se preserva el derecho del posible portador a no querer saber si ha heredado la enfermedad.
Leer más
¿Se puede prevenir la transmisión de la enfermedad de Huntington?
Sí. La corea de Huntington se puede evitar consiguiendo el embarazo mediante fecundación in vitro (FIV) con diagnóstico genético preimplantacional (DGP).
Gracias al DGP, únicamente se transferirán aquellos embriones sanos que no contengan la mutación del gen causante de la enfermedad de Huntington (HTT).
¿Qué tipo de herencia tiene la corea de Huntington?
La enfermedad o corea de Huntington, en honor al nombre de su descubridor, es una patología genética con herencia autosómica dominante. Esto significa que únicamente se requiere una copia del gen alterado para heredar la enfermedad.
Lecturas recomendadas
Si deseas obtener información acerca de otras enfermedades que se pueden evitar mediante un DGP, puedes visitar el siguiente artículo: ¿Qué son las enfermedades monogénicas? - Evitarlas con DGP.
Además, si lo que te gustaría es saber más acerca de esta técnica complementaria de reproducción asistida para analizar los embriones, entonces te invitamos a acceder al siguiente enlace: ¿Qué es el diagnóstico genético preimplantacional o DGP?
Hacemos un gran esfuerzo para ofrecerte información de máxima calidad.
🙏 Por favor, comparte este artículo si te ha gustado. 💜💜 ¡Nos ayudas a seguir!
Bibliografía
Amy Kim, Kathryn Lalonde, Aaron Truesdell, Priscilla Gomes Welter, Patricia S Brocardo, Tatiana R Rosenstock, Joana Gil-Mohapel. New Avenues for the Treatment of Huntington's Disease. Int J Mol Sci. 2021 Aug 4;22(16):8363. doi: 10.3390/ijms22168363 (Ver)
Eric Fields, Erik Vaughan, Deepika Tripu, Isabelle Lim, Katherine Shrout, Jessica Conway, Nicole Salib, Yubin Lee, Akash Dhamsania, Michael Jacobsen, Ashley Woo, Huijing Xue, Kan Cao. Gene targeting techniques for Huntington's disease. Ageing Res Rev. 2021 Sep;70:101385. doi: 10.1016/j.arr.2021.101385. Epub 2021 Jun 5 (Ver)
Jae Wook Hyeon, Albert H Kim, Hiroko Yano. Epigenetic regulation in Huntington's disease. Neurochem Int. 2021 Sep;148:105074. doi: 10.1016/j.neuint.2021.105074 (Ver)
James F Gusella, Jong-Min Lee, Marcy E MacDonald. Huntington's disease: nearly four decades of human molecular genetics. Hum Mol Genet. 2021 Oct 1;30(R2):R254-R263. doi: 10.1093/hmg/ddab170 (Ver)
Monia Barnat, Mariacristina Capizzi, Esther Aparicio, Susana Boluda, Doris Wennagel, Radhia Kacher, Rayane Kassem, Sophie Lenoir, Fabienne Agasse, Barbara Y Braz, Jeh-Ping Liu, Julien Ighil, Aude Tessier, Scott O Zeitlin, Charles Duyckaerts, Marc Dommergues, Alexandra Durr, Sandrine Humbert. Huntington's disease alters human neurodevelopment. Science. 2020 Aug 14;369(6505):787-793. doi: 10.1126/science.aax3338. Epub 2020 Jul 16 (Ver)
Nicholas E Karagas, Natalia Pessoa Rocha, Erin Furr Stimming. Irritability in Huntington's Disease. J Huntingtons Dis. 2020;9(2):107-113. doi: 10.3233/JHD-200397 (Ver)
Nóra Zsindely, Fruzsina Siági, László Bodai. DNA Methylation in Huntington's Disease. Int J Mol Sci. 2021 Nov 25;22(23):12736. doi: 10.3390/ijms222312736 (Ver)
Sarah J Tabrizi, Carlos Estevez-Fraga, Willeke M C van Roon-Mom, Michael D Flower, Rachael I Scahill, Edward J Wild, Ignacio Muñoz-Sanjuan, Cristina Sampaio, Anne E Rosser, Blair R Leavitt. Potential disease-modifying therapies for Huntington's disease: lessons learned and future opportunities. Lancet Neurol. 2022 Jul;21(7):645-658. doi: 10.1016/S1474-4422(22)00121-1 (Ver)
Sarah J Tabrizi, Rhia Ghosh, Blair R Leavitt. Huntingtin Lowering Strategies for Disease Modification in Huntington's Disease. Neuron. 2019 Mar 6;101(5):801-819. doi: 10.1016/j.neuron.2019.01.039 (Ver)
Sigita Lesinskienė, Darja Rojaka, Rūta Praninskienė, Aušra Morkūnienė, Aušra Matulevičienė, Algirdas Utkus. Juvenile Huntington's disease: two case reports and a review of the literature. J Med Case Rep. 2020 Oct 1;14(1):173. doi: 10.1186/s13256-020-02494-7 (Ver)
T Maiuri, C E Suart, C L K Hung, K J Graham, C A Barba Bazan, R Truant. DNA Damage Repair in Huntington's Disease and Other Neurodegenerative Diseases. Neurotherapeutics. 2019 Oct;16(4):948-956. doi: 10.1007/s13311-019-00768-7 (Ver)
Preguntas de los usuarios: '¿La enfermedad de Huntington afecta a la fertilidad?', '¿Puedo ser madre si sufro la enfermedad de Huntington?', '¿Se puede prevenir la transmisión de la enfermedad de Huntington?' y '¿Qué tipo de herencia tiene la corea de Huntington?'.
Autores y colaboradores

Además, el doctor ha participado en varias publicaciones científicas y ponencia, aparte de realizar cursos de formación complementaria. Más sobre Dr. John Peay Pinacho


Más sobre Dr. Sergio Rogel Cayetano
